Adsorption and Diffusion in Nanoporous materials

Dr. David Dubbeldam
Computation Chemistry Group since 2009. Lead-author of the iRASPA and RASPA molecular simulations codes.
Ordered crystalline porous materials offer the potential for selective adsorption by exploiting differences in molecular configurations. Zeolites are readily available, very stable, and cheap. A zeolite should have the right combination of high adsorption selectivity, combined with an adequate capacity for use in traditionally used fixed-bed devices. Recently, new classes of nanoporous materials have been designed that have good stability, high void volumes, and well-defined tailorable cavities of uniform size. Example are metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and zeolitic imidazolate frameworks (ZIFs). These novel materials possess almost unlimited structural variety because of the many combinations of building blocks that can be imagined. MOFs are synthesized under solvo- or hydrothermal conditions in the presence of a base. During synthesis the building blocks self-assemble into crystalline materials that, after evacuation, can find applications in adsorption separations, air purification, gas storage, chemical sensing, and catalysis.
Ordered crystalline porous materials offer the potential for selective adsorption by exploiting differences in molecular configurations. Zeolites are readily available, very stable, and cheap. A zeolite should have the right combination of high adsorption selectivity, combined with an adequate capacity for use in traditionally used fixed-bed devices. Recently, new classes of nanoporous materials have been designed that have good stability, high void volumes, and well-defined tailorable cavities of uniform size. Example are metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and zeolitic imidazolate frameworks (ZIFs). These novel materials possess almost unlimited structural variety because of the many combinations of building blocks that can be imagined. MOFs are synthesized under solvo- or hydrothermal conditions in the presence of a base. During synthesis the building blocks self-assemble into crystalline materials that, after evacuation, can find applications in adsorption separations, air purification, gas storage, chemical sensing, and catalysis.
Regenerative adsorber systems can operate continuously by using:
- multiple fixed beds of an adsorbent;
- fluidized bed contactors with separate adsorption and desorption vessels; or
- rotary bed adsorbents that cycle continuously between adsorption and desorption operations.
Pressure swing adsorption (PSA) is an industrial separation technique at atmospheric temperature and high pressure. In practice a batch setup with various vessels in parallel is used; some are in adsorption mode, while others are in regeneration mode. In pressure swing the regeneration is done at low pressure and temperature, while thermal swing adsorption uses high temperature and pressure. PSA is less sensitive to diffusional effects and largely dominated by adsorption. This is in contrast to membrane technology, where the difference in mobility of species leads to separation. The difficulty of screening lies in determining what most suitable means. The economics of PSA is crucially dependent on adsorption selectivity. High adsorption selectivity leads to smaller equipment volumes and lower capital and energy requirements. However, high selectivity means a strong affinity of the most adsorbing species and therefore higher regeneration costs. Besides selectivity, it is also the capacity of the material that determines the efficiency of a separation device. In pressure swing adsorbers, high adsorption capacities are desirable because they result in lower frequencies in the PSA regeneration cycles. Other crucially important factors are cost of the material and stability (especially temperature and water stability); once the MOF is evacuated it is likely to become air and moisture sensitive. A reliable screening methodology should take all these considerations into account.
Soft Matter
Soft materials are ubiquitous in everyday life, including products such as food and cosmetics, but also biological and bioinspried materials in general. Chraracterise by energy scales that are influenced by thermal motion, they show complex features over a wide range of length and time scales. Computational soft matter research aims to understand and predicted the emergent complexity of soft-matter systems by means of simulations, theory and modelling. This interdisciplinary discipline requires a combination of advanced computational methods, theoretical physics and chemistry, as well as more recent data-driven methods. The computational soft matter laboratory has been recently founded to simulate the interdisciplinary effort .
Self assembly

Dr. Ir. Ioana M. ILIE
Computational Chemistry Group.
The self-assembly of proteins into ordered aggregates on relevant timescales is computationally very demanding at atomistic resolution. Hence, the thermodynamics and kinetics of aggregate formation are difficult to be explored. Understanding the accumulation of proteins and their relationship to triggering various biological responses are crucial elements in generating reliable predictions and explaining experimental observations.
Patchy particles are well suited to model self-assembly of proteins into ordered or amorphous aggregates. Combined with Brownian dynamics they provide insight into the dynamics of the assembly process. Proteins are modelled as single nanometer sized particles decorated with interaction patches. Additionally, the particles are able to change shape and interaction parameters in response to the environment. In collaboration with the Organic Chemistry and Physical Chemistry group at the Department of Biomolecular Sciences of Wageningen University, we explore the aggregation of polypeptides associated with neurodegenerative diseases (e.g. amyloid-beta for Alzheimer’s disease, alpha-synuclein for Parkinson’s disease) by developing novel models and using advances simulation methods.
Active Soft Matter

Prof. dr. Peter Bolhuis
Computational Chemistry Group.
Structural architectures in living cells, such as the cytoskeleton in muscle or plant tissue, are both viscoelastic and active, i.e. undergo continuous injection of energy, leading to remarkable collective, non-equilibrium properties. The thermodynamics, kinetics and statistical mechanics of such systems are far from understood. While active fluids (swarms, swimmers) have seen an explosion of interest recently, their solid (elastic) counterparts remain completely unexplored and understanding such phenomena remains one of the grand challenges of modern statistical physics.
Patchy particles provide a model system for exploring complex molecular structures with high controllability. Patchy particles are micron-sized colloidal particles dressed with patches that are able to form bonds under specific solvent compositions and temperatures. While patchy particles are big enough to be observed in experiment under e.g. a confocal microscope, they are small enough to exhibit similar statistical behavior as atoms and molecules. In addition, patchy particles are a mesoscopic structural analogue of carbon atoms. Divalent patchy particles make linear bonds like sp hybridized carbon atoms and act as monomers forming chains as shown in the background. Tetravalent patchy particles make tetrahedral bonds and may form rings with a half-chair or envelope conformation like cyclopentane. In collaboration with the experimental Soft Matter group we explore active patchy particle architectures, by using advanced simulation methods, including path sampling.
Design of soft matter

Dr. Alberto Pérez de Alba Ortíz
Computational Chemistry Group.
Alberto Pérez de Alba Ortíz focuses on the development of computational frameworks for bottom-up design of self-assembling materials with tailor-made properties. To this end, his group uses evolutionary, Bayesian and active learning models to predict desirable properties and thus decide which combination of design parameters is more suitable to try next, either in simulation or experiments. The data collected during these iterative design also serves as training for future generative and predictive machine learning models. Current projects are focused on time-dependent protocols for colloidal self-assembly and responsive mechanical properties in polymers, as well as self-assembly of two-component quasicrystals and zeolitic phases. More over, by means of mimicking indirect observations, these design strategies can explain measurements of matter that is impossible to reach by experiments, e.g., the phase behavior of iron at our planet’s inner-core.
Computational Polymer Chemistry and Science for Arts

Prof. dr. P. Iedema
Polymer Chemistry and Computational Chemistry Group.
The group aims at fundamental understanding of chemical and physical processes in a wide range of polymer systems, from oil paint layers to industrially produced compounds, covering polymerization and long-term degradation. We take a macroscopic perspective using Chemical Engineering modeling tools to describe chemical kinetics and transport phenomena. The group collaborates with industries like Canon Production Printing Netherlands in Venlo, Sabic in Geleen, BASF in Ludwigshafen, but also with the Rijksmuseum and the Netherlands Institute for Conservation, Art and Science, NICAS. The Science for Arts activities of the group have an experimental component described elsewhere (https://hims.uva.nl/research/research-topics/chemistry-for-conservation-and-art).
Polymer synthesis, processing and degradation
This aims at predicting end use properties of polymers by elucidating microstructure as characterized by chain length distribution, type and numbers of branching, copolymer composition. Typical results nowadays attainable are radius of gyration and rheological properties predicted for industrial polymers like polyolefins and acrylates. We develop mathematical tools to deal with the combinatorial explosion of wide ranges of molecular sizes, degrees of branching, copolymer compositions.
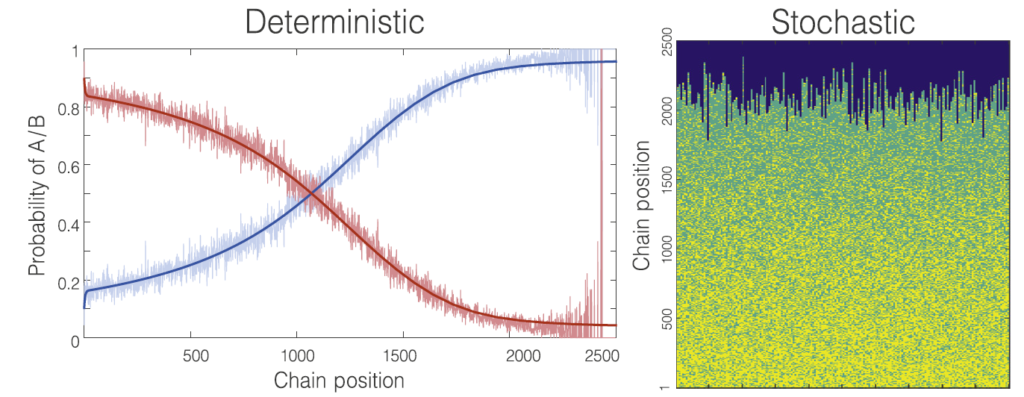
Polymer networks
Modelling stochastic polymer networks is challenging since they are complex systems created starting from a non-percolating phase (a ‘liquid’ sol) and ending in a percolating phase (a ‘solid’ gel). While a classical molecular dynamics approach works fine for simple networks with a repetitive pattern, it often collapses with stochastic networks because of the inability of modelling in a reasonable time. Our approach to model polymer networks views the assembly of molecular networks from the perspective of evolving graph topology. This describes randomly fluctuating polymerization that from a very simple topology to a complicated interconnected structure.
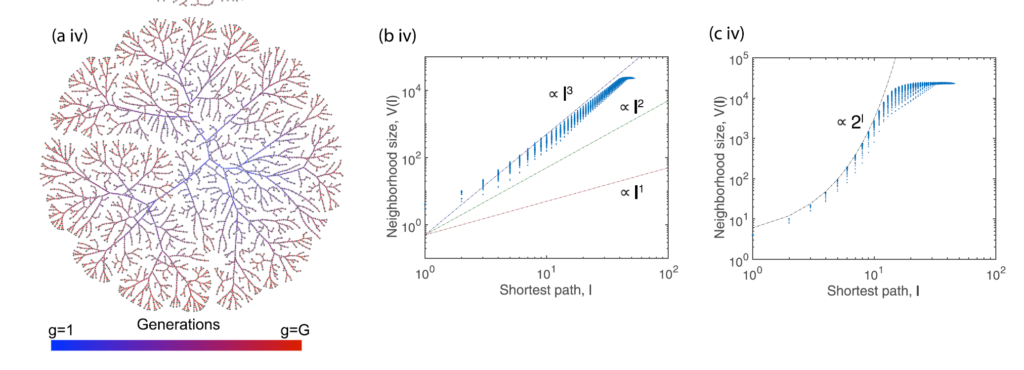
Effect of volume growth model on network structure (Schamboeck, V., Kryven, I. & Iedema, P. D. Effect of volume growth on the percolation threshold in random directed acyclic graphs with a given degree distribution. Phys. Rev. E 101, (2020)).
Transport Phenomena
Swelling and diffusion of polymer films are studied in various applications. During the creation of thin acrylate films the process is disturbed by oxygen inhibition. Old paintings are cleaned using solvents, but the complex, multi-component diffusion and swelling of paint lay ers, possibly accompanied by metal-soap reactions are not well understood. Compartment modeling in combination with parameter estimation techniques is developed to find kinetic and diffusion parameters from experiments.
Determination of polymer concentration-dependent diffusion coefficient from ATR-FTIR data (Baij, L., Hermans, J. J., Keune, K. & Iedema, P. D. Time-Dependent ATR-FTIR Spectroscopic Studies on Solvent Diffusion and Film Swelling in Oil Paint Model Systems. Macromolecules 51, 7134–7144 (2018)).
Automated reaction network generation
We study computer algorithms as an aid for the discovery of complex reaction networks. Typically, in this class of problems, it is the complexity of data augmented by the combinatorial explosion of the number of choices that exceeds human performance, which is also the case for the reaction kinetics of, for instance, linseed oil polymerization, which forms the binding medium of oil paint. We develop an automated procedure using the hierarchy present in the chemical notation as a formal language operating with graphs as sentences and by viewing the reaction mechanisms as a graph grammar to build an algorithm that constructs the network of organic chemistry reactions.
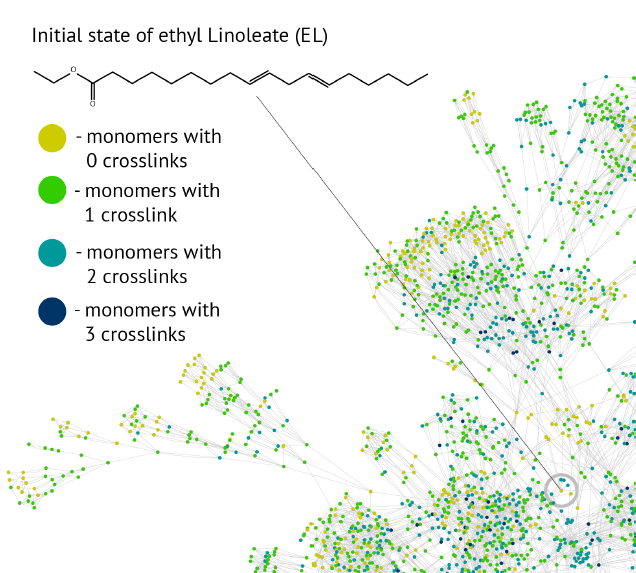
Part of reaction network of Ethyl Linoleate (Orlova, Y., Gambardella, A. A., Harmon, R. E., Kryven, I. & Iedema, P. D. Finite representation of reaction kinetics in unbounded biopolymer structures. Chem. Eng. J. 126485 (2020)>