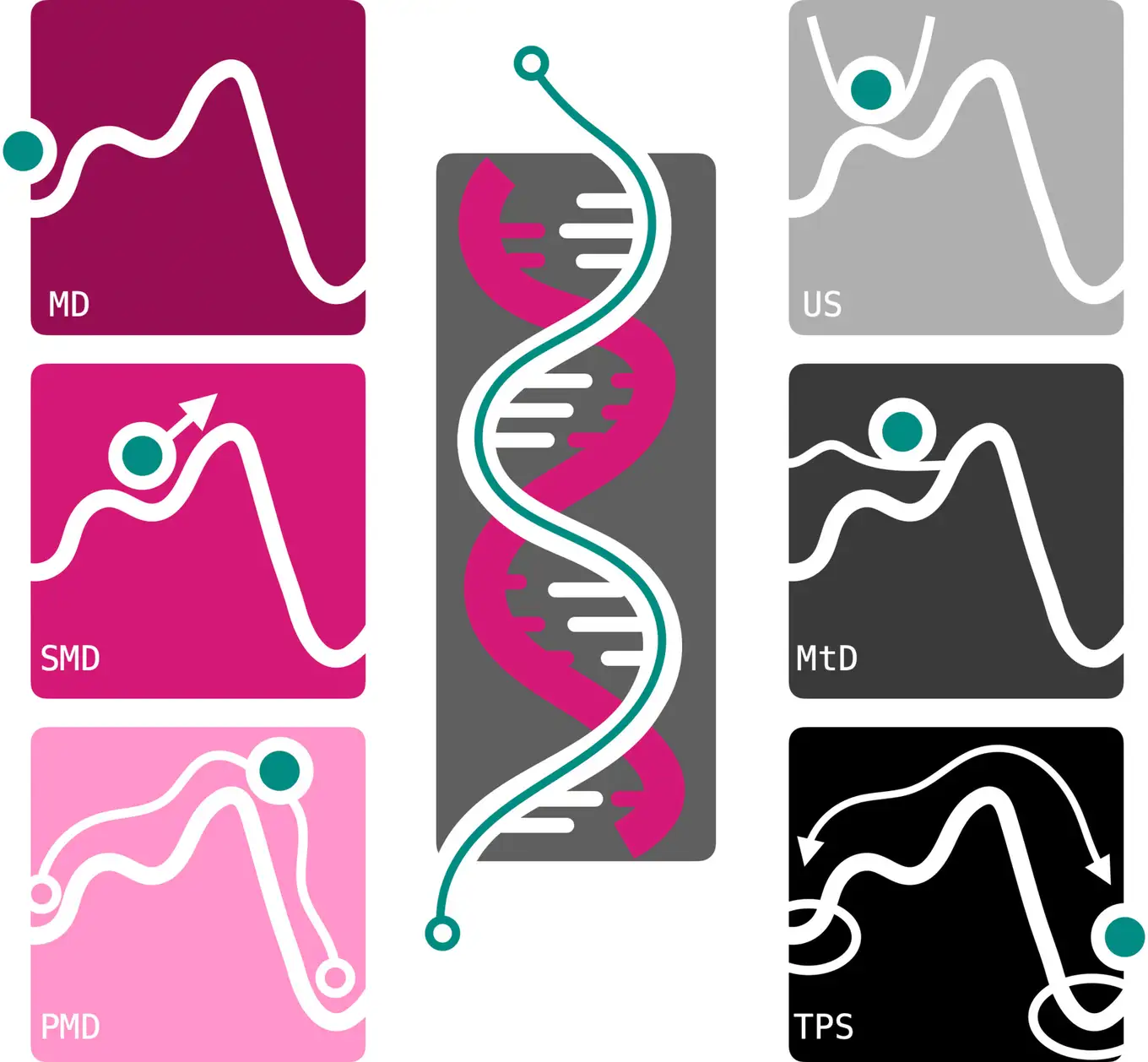
In a review paper that has been recently published in WIREs Computational Molecular Science, researchers of the Computational Science group at the Van ‘t Hoff Institute for Molecular Sciences describe successful approaches for simulating molecular systems involving DNA. They show how these can be combined in several ways to tackle complex DNA transitions, integrating information about states and pathways.
All-atom molecular dynamics (MD) is a powerful simulation method to obtain insights into molecular processes at atomic detail. By generating ‘molecular movies’, MD simulations make processes accessible that cannot be observed in experiments. As MD is a numerical method, many calculations are required to obtain sufficient accuracy. For many processes involving DNA this poses a huge challenge. In this review, several simulation strategies are demonstrated that yield accurate results with high detail and within a reasonable time.
Abstract of the paper
Molecular dynamics (MD) simulations can provide detailed insights into complex molecular systems, such as DNA, at high resolution in space and time. Using current computer architectures, time scales of tens of microseconds are feasible with contemporary all-atom force fields. However, these timescales are insufficient to accurately characterize large conformational transitions in DNA and compare calculations to experimental data. This review discusses the advantages and drawbacks of two simulation approaches to overcome the timescale challenge. The first approach is based on adding biasing potentials to the system to drive transitions. Umbrella sampling, steered MD, and metadynamics are examples of these methods. A key challenge of such methods is the necessity of selecting one or a few efficient coordinates, commonly referred to as collective variables (CVs), along which to apply the biasing potential. The path-metadynamics methodology addresses this issue by finding the optimal route(s) between states in a multi-dimensional CV space. The second strategy is path sampling, which focuses MD simulations on the transitions. The assumption is that even though transitions between states are rare, they are generally fast. Stopping the simulations as soon as they reach a stable state can significantly increase simulation efficiency. We introduce these methods on the two-dimensional Müller–Brown potential. DNA applications are featured for two different processes: the Watson–Crick–Franklin to Hoogsteen transition in adenine–thymine base pairs and the binding of a DNA-binding protein domain to DNA.
Paper details
Bernadette Mohr, Thor van Heesch, Alberto Pérez de Alba Ortíz, Jocelyne Vreede: Enhanced sampling strategies for molecular simulation of DNA.WIREs Computational Molecular Science Volume 14, Issue 2 e1712